
Perspectives on Allele-Specific Expression
2021年4月28日 Annual Review of Biomedical Data Science 在线发表关于等位基因特异性表达的综述,其中详细介绍最近针对Allele-Specific Expression(ASE)的方法学发展和对ASE的分析结果,其主要集中在人类数据上。

Abstract
二倍体物种对群体遗传学和遗传性疾病的研究具有深远的影响。虽然人类基因组中的大多数基因都有两个拷贝,但在一个特定的个体中,它们不一定都是活跃的或在同一水平上。基因组印记,导致有利于父系或母系等位基因的排他性或偏向性表达,现在认为影响了数百个人类基因。更多的基因显示出基因拷贝的不平衡表达,这是由于顺式作用的基因变异扰乱基因表达。通过对大量个体和组织类型的RNA测序产生的数据的可用性产生了前所未有的机会来评估基因变异对等位基因不平衡的贡献。本综述通过分析这些数据所获得的关于基因变异对等位基因表达不平衡的贡献程度,基因表达不平衡的分析工具和统计模型,以及所获得的结果来揭示关于改变基因表达的遗传变异对复杂人类疾病和表型的贡献。
1. INTRODUCTION
当二倍体(或多倍体)生物体内某个基因座的等位基因的状态或活性有差异时,就会出现等位基因不平衡。许多关于等位基因不平衡的研究都集中在mRNA丰度的差异上,将其称为 "等位基因表达不平衡"。等位基因之间mRNA丰度的不平衡被称为 "等位基因特异性表达"(allele-specific expression)(ASE)。这个术语经常被用来指基因表达的不平衡,而不考虑表达的差异是由于遗传变异还是表观遗传效应,如基因组印记或随机单倍体表达(RMAE)。然而,由于它暗示了产生于等位基因本身的效应,所以 "等位基因特异性表达 "这一术语应保留给具有遗传起源的不平衡,并在此采用这一用法。当等位基因之间表达差异的原因不明确时,使用 "等位基因表达不平衡"(allelic expression imbalance)这一术语。同样,用 "等位基因特异性不平衡"(allelic imbalance)来指代等位基因之间在染色质状态、表达水平或剪切体相对丰度方面的任何差异,而当这些差异起源于遗传时,则用 "等位基因表达不平衡"(Allele-specific imbalance)。
Allelic imbalance: 一个基因座上的等位基因之间的染色质状态或mRNA或蛋白质水平的差异
Allelic expression imbalance: 导致mRNA水平差异的等位基因不平衡,从一个等位基因完全表达的完全不平衡到等位基因之间表达的细微差异
Allele-specific imbalance: 由于等位基因之间的遗传差异,等位基因之间的染色质状态或mRNA或蛋白质水平的差异。
Allele-specific expression: 源于遗传的等位基因表达不平衡,即由于等位基因序列之间的差异导致等位基因之间表达水平的差异
ASE与表达数量性状位点(expression quantitative trait loci)(eQTLs)有密切联系,eQTLs是对基因表达有影响的遗传位点。eQTLs可以起顺式(cis)作用,影响同一染色体上一个基因的表达,通常位于该位点附近;也可以用于反式(trans)作用,在这种情况下,eQTL和受影响的基因可能是不相关的。当它起顺式作用时,eQTL通常会导致杂合子的替代等位基因(alternative alleles)之间的不平衡。因此,等位基因不平衡常常被用来鉴定cis-eQTLs的。ASE在检测调控性变异,特别是罕见性突变方面具有突出作用。调控性变异对人类表型和复杂疾病易感性变异上具有重要作用,这使得人们对这种现象越来越感兴趣。这反过来又推动了检测ASE本身和利用ASE来推断调控变异及其对基因表达的影响的方法的激增。
本文回顾了最近的方法学发展和对ASE的分析结果,主要集中在人类数据上。首先概述了等位基因不平衡的类型,并回顾了其他类型的不平衡可能导致ASE的机制。讨论了一系列的统计模型和计算流程,这些模型和流程已经被开发出来,用于从高通量测序数据中识别等位基因不平衡,并利用ASE来识别eQTLs和优先考虑可能是人类疾病因果关系的基因组变异。回顾了人类样本中ASE的估计流行率,并讨论了ASE对遗传病渗透率的影响,以及它对发现全基因组关联研究(GWAS)中发现的与表型关联的因果变异的潜力。
2. MECHANISMS OF ALLELIC IMBALANCE AND ALLELE-SPECIFIC EXPRESSION
遗传变异可对染色质结构、基因转录和转录后调控产生影响,包括mRNA剪切、microRNA(miRNA)结合和mRNA翻译(图1)。在许多情况下,这些变异可以影响相关等位基因的表达水平,导致ASE,以及其他可测量的等位基因不平衡的形式。例如,改变转录因子结合位点的遗传变异可导致转录因子结合的不平衡,并通过改变转录启动的速度而导致ASE的发生。同样,mRNA剪切中的等位基因特异性不平衡,其本身可影响基因功能并揭示剪切调控变异,可通过改变翻译框架和诱导无意义介导的衰变而导致ASE。下面将讨论一些导致等位基因特异性不平衡的主要机制,强调其中一些机制引起ASE的可能性。
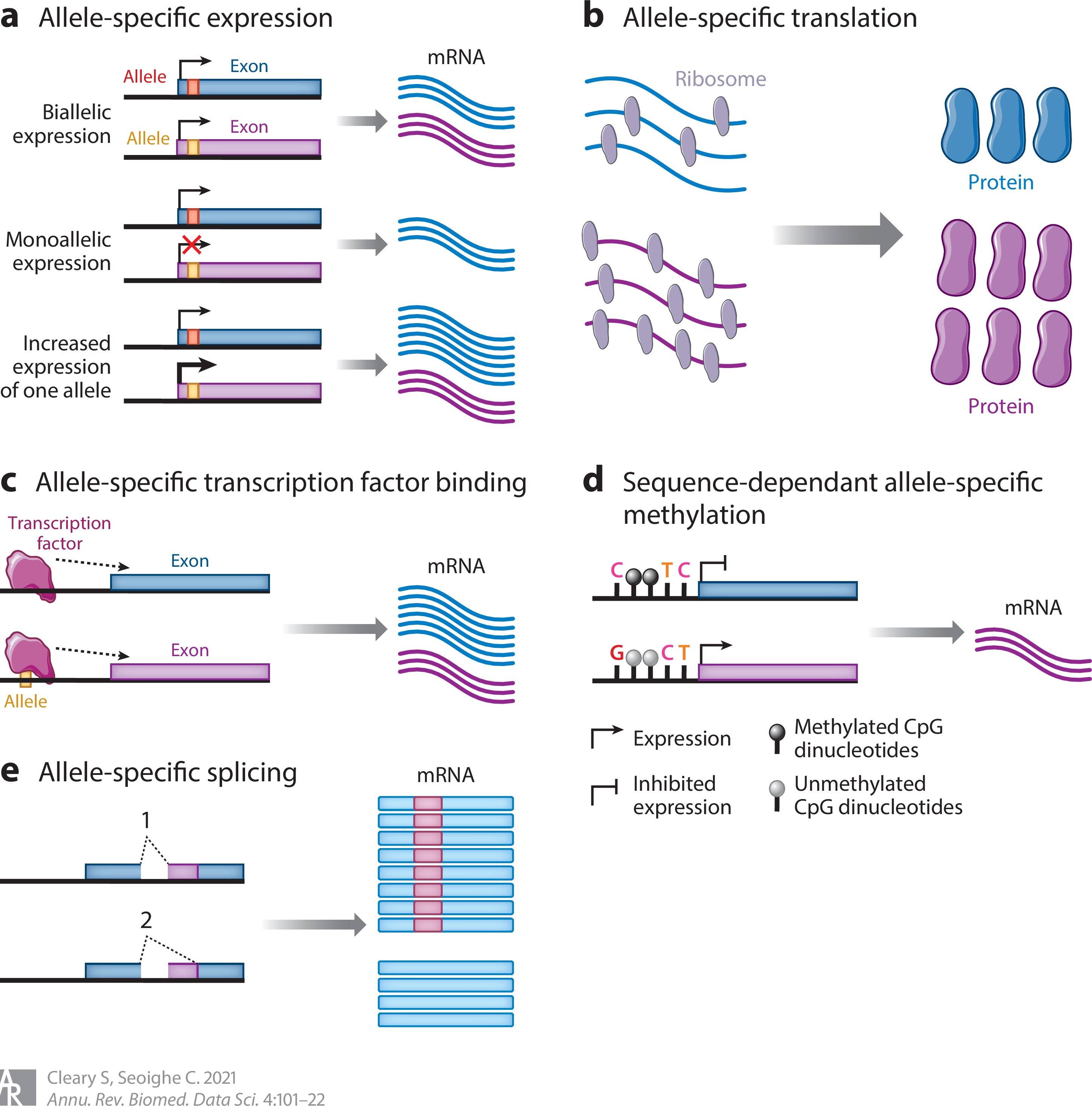
(a) 等位基因表达不平衡:显示了三种情况:两个等位基因的表达相同(顶部),一个等位基因的单独表达(中间),以及一个等位基因的较高表达(底部)。
(b) 翻译上的等位基因不平衡:遗传变异可以改变mRNA翻译的速度,导致等位基因之间核糖体的占用水平不同。
(c) 转录因子结合的不平衡:在所示的例子中,一个序列变异 降低了转录因子的结合亲和力,导致了等位基因的特异性表达。
(d) DNA甲基化的不平衡:甲基化抑制了一个等位基因的表达。如果甲基化的差异是由顺式作用的遗传变异导致的,它可以导致等位基因特异性表达。
(e) 等位基因特异性剪接:变异导致两个等位基因产生不同的剪切体。
2.1. Transcriptional Regulation
非编码区的杂合单核苷酸多态性(SNPs)可能通过影响转录因子的结合亲和力或导致替代等位基因之间染色质状态的差异而对转录率产生下游影响,从而成为ASE的原因。染色质修饰在非遗传性的等位基因不平衡中也起着关键作用(包括基因组印记和X染色体失活)。遗传和表观遗传效应之间也可以通过等位基因特异性DNA甲基化发生相互作用。
2.1.1. Transcription factor binding.
具有杂合SNP的等位基因对转录因子有不同的亲和力,导致转录因子占有的等位基因不平衡,每个等位基因的转录率不同(图1c)。对等位基因特异性转录因子结合的分析在理解非编码DNA如何影响基因表达和引起疾病表型方面发挥了重要作用。为了全面剖析结合能力改变的影响,应该确定致病基因调控变异,与之结合的转录因子和靶基因。
2.1.2. Chromatin accessibility.
表观遗传标记,如DNA甲基化和组蛋白修饰,可以跨细胞世代遗传,产生表达相同等位基因的不同细胞群。印记和X染色体失活是由于表观遗传效应而不是等位基因之间的遗传差异。等位基因之间的遗传差异引起的表观遗传差异也可以通过一个称为序列依赖的等位基因特异性甲基化的过程导致ASE(图1d)。这可以以组织和个体的方式影响非印记的常染色体基因。约有5%的CpG位点显示出DNA甲基化的大量(>30%)不平衡的证据。组蛋白修饰比CpG甲基化更复杂,H3和H4组蛋白尾部的不同氨基酸可能有数百种不同类型的组蛋白尾部修饰,包括乙酰化、甲基化、磷酸化、泛素化、苏木酰化和ADP-核糖化。遗传变异对基因表达的影响在许多情况下是由它们对染色质修饰的影响所介导的。染色质可及性的差异也可由等位基因特异性转录因子的结合导致,并可能对复杂的疾病做出重大贡献。
2.2. Posttranscriptional Mechanisms
在基因转录后,有多种调节机制作用于基因。由于这些机制通常具有序列特异性,它们的作用在不同的等位基因之间可能有所不同,从而导致等位基因的不平衡。
2.2.1. Nonsense-mediated decay and alternative splicing.
无义突变介导的衰变(NMD)是一种关键的细胞质量控制机制,其结果是消除携带过早终止密码子(PTC)的mRNA,这些密码子可能导致畸形的蛋白质。这一过程发生在细胞质中,与翻译的终止和mRNA的降解有关。NMD在控制mRNA表达水平方面也起着作用,有助于调节大量的人类基因。一个杂合的SNP,其中一个等位基因导致的PTC可以导致来自该等位基因的mRNA的降解,从而导致ASE。遗传变异可以通过改变转录本中的剪切信号来影响mRNA的剪切。这种突变可以发生在剪切供体或受体位点内或附近,在分支点周围,或在外显子、内含子、增强子或抑制子位点。对剪切的常见影响包括外显子跳跃、内含子保留、3′或5′外显子末端交替、以及相互排斥的外显子。由于它们在顺式中起作用,转录的剪切突变通常会导致等位基因特异性剪切。当改变mRNA剪切的突变引入一个内源终止密码子时(例如,通过跳过编码区的一个外显子,该外显子的长度不是三个核苷酸的倍数),它可以触发针对受影响等位基因的NMD。这导致造成错误剪切的等位基因的成熟mRNA的丰度低于野生型等位基因,从而导致ASE。即使NMD没有被触发,由影响剪切的遗传变异引起的蛋白质剪切体之间的差异也会产生重要的功能影响。
2.2.2. Variants affecting mRNA binding sites.
RNA结合蛋白(RBPs)在转录后的基因调控中发挥着作用,它以一种序列特异性的方式与RNA结合,调控结合的RNA的命运。mRNA上的遗传变异可以破坏RBPs与mRNA的相互作用,导致RNA结合的等位基因不平衡,并可能导致mRNA定位或翻译的ASE或等位基因变异。在ENCODE(DNA元件百科全书)的eCLIP-Seq(增强型交联和免疫沉淀测序)数据中,应用一种检测RNA结合的等位基因不平衡的方法,发现了改变mRNA剪切和基因表达水平的基因组变异,说明了等位基因特异性RNA结合导致ASE的潜力。这些非编码RNA本身可以显示其表达的等位基因特异性不平衡,并在它们调节的基因中诱发ASE。与蛋白质编码的mRNAs相比,lncRNAs在其表达中表现出更大程度的等位基因不平衡。miRNA与其目标mRNA的相互作用可受mRNA中与miRNA互补的位点内SNP的影响,同样,这可能会导致ASE。
2.3. Translational Mechanisms
基因组变异产生或破坏了5′非翻译区的上游起始密码子,改变了mRNA的二级结构,影响了翻译起始位点或附近的序列基序,或产生了新的mRNA异构体,这些都会影响mRNA的翻译。由于这些变异都是顺式作用,它们可以导致mRNA翻译速度的等位基因不平衡,以及在某些情况下改变所产生的蛋白质产物。Li等人将核糖体相关和非核糖体相关RNA的比例视为定量性状,确定了推断与人类淋巴细胞系中mRNA翻译率有显著关系的SNP。最近的一项研究使用翻译核糖体亲和纯化来确定与核糖体占有率改变有关的遗传变异,并发现有证据表明上游开放阅读框、miRNA结合位点和poly-A信号的序列变异导致小鼠星形细胞中1-2%的转录物的翻译效率变化。翻译率的等位基因变异可以造成来自两个等位基因的蛋白质丰度的不平衡,即使在mRNA水平上没有ASE。等位基因之间RNA编辑的不平衡也会导致蛋白质水平的差异,在这种情况下,氨基酸序列会出现意想不到的变化。在全基因组范围内寻找等位基因特异性的RNA编辑,发现了同义SNP的例子,导致附近的非同义变化是由RNA编辑引起。
3. ALLELIC IMBALANCE ANALYSIS
3.1. Experimental Design Considerations and Computational Pipelines
从高通量测序数据中分析等位基因不平衡和ASE,通常需要生成测序reads以及mapping到每个等位基因的序列的reads count。生成这一数据涉及多个步骤,详见下文,其中每个步骤都与潜在的偏差和混杂因素有关。有几个高效和可扩展的流程可用于这些任务,如AlleWorkbench、WASP、CloudASM和ALEA。下面的讨论主要集中在从RNA测序(RNA-seq)数据中推断等位基因表达不平衡,但类似的方法也可以适用于其他类型的等位基因不平衡。
3.1.1. Sequencing.
为了有足够的能力来区分替代等位基因的表达水平,等位基因不平衡的分析需要比典型的RNA-seq实验中产生的更高的覆盖度,重点是总的表达分析。通常采用30个reads跨越感兴趣的位置的阈值来推断单个杂合子位点的等位基因不平衡。这可以限制具有足够覆盖度的基因数量,以检测等位基因不平衡。例如,在Geuvadis的研究中,每个样本的mapping reads的中位数为55 million,符合这个阈值的基因中位数只有3000个。这受到样本杂合度以及测序深度的影响,而且没有考虑mapping到单倍型而不是单个杂合SNP的可能性。基因型-组织表达(GTEx)项目进行RNA测序的中位深度为83 million reads,使用基于单倍型的方法,可以测试更多比例的基因的不平衡性。在 bulk RNA-seq实验的情况下,只有有丝分裂稳定的等位基因不平衡(即来自原始细胞的所有子细胞共享一个等位基因的表达模式)可以被检测到。单细胞RNA-seq可以提供动态不平衡的信息,这种不平衡随着时间的推移而变化,例如,转录活性的爆发。这可能导致等位基因不平衡的模式在连续的细胞世代中不稳定。
3.1.2. Alignment and removal of polymerase chain reaction duplicates.
分析等位基因不平衡的软件流程的第一步是将序列reads align到参考基因组或转录组。在alignment或mapping中的错误会对获得的结果产生重大影响。mapping错误(将reads mapping到错误的位置或未能map到read)可能会在含有杂合子SNP的替代等位基因的reads上发生,频率更高,导致等位基因不平衡的假阳性信号。已经提出了一些策略来减少序列比对的偏差。这些策略包括使用masked reference;使用自己设计的二倍体基因组或转录组;用于比对的单倍型基因组;SNP-tolerant的mapping工具,如GSNAP,STAR-WASP,ASE-lux和SNP-omatic;以及使用remapping策略的方法,如WASP。据报道,将reads与包括遗传变异在内的二倍体转录组进行比对的方法可提高ASE的估计值。
基因水平上的不平衡推断提供了一个不完整的结果,因为剪切异构体之间可能存在不同程度的不平衡,具有功能影响。也有报道说,当剪切异构体被忽略时,在基因水平上推断ASE是有偏差的。对于推断异构体水平的表达不平衡,序列reads必须被mapping到它们所产生的异构体及其等位基因。对于reads map到转录本和等位基因,可能存在模糊性,特别是由于许多reads不与杂合子SNP或区分不同等位基因的转录本区域重叠。这一点已经通过使用分级期望最大化策略对reads进行加权分配来解决,据报道,这导致了一般等位基因不平衡推断的改进,包括在剪切异构体水平。
在准备测序文库的过程中,聚合酶链反应(PCR)扩增步骤可能导致同一互补的DNA片段被多次测序。这导致了具有相同mapping坐标的sequence reads。虽然识别这些重复的reads并将其删除是很简单的,但由于高表达基因信息的丢失,一般不建议对RNAseq数据进行这种处理。然而,对等位基因不平衡的统计试验往往对重复reads的存在并不robust,因此,在分析等位基因不平衡之前,应删除潜在的PCR重复reads。许多去除重复reads的工具保留了具有最佳mapping score的reads,但是对于等位基因不平衡的分析,必须使用诸如WASP这样的工具来随机选择保留的reads,以避免有利于参考等位基因的mapping偏差。
3.1.3. Genotyping and haplotype phasing.
等位基因特异性read counts的产生需要在目标特征(基因、转录因子结合位点等)内至少有一个杂合的SNP。杂合的SNP可以用基因分型矩阵或基因组DNA测序来单独鉴定。或者,杂合子SNPs可以从map到感兴趣的特征的reads中推断出来。例如,在ASE的情况下,基因型可以从RNA-seq reads中推断出来。然而,这有一个风险,即显示极端不平衡的特征可能被误称为同源位点,导致等位基因不平衡的推断出现假阴性。相反,测序错误、转录错误,甚至是罕见的体细胞突变,导致在生殖细胞系中同源的位点被错误地称为杂合子,都会导致等位基因不平衡推断的假阳性。在基因组DNA上进行基因分型时也可能发生错误。在这种情况下,同源位点被错误地认为杂合子会导致等位基因不平衡的假阳性推断。最近分析等位基因不平衡的方法考虑到了基因分型的不确定性。
准确的SNP分型数据支持等位基因不平衡的推断,通过允许reads mapping到跨越多个杂合SNP的单倍型。序列reads中包含的信息可用于此目的,当有long reads数据时,可获得更高的准确性。从群体分型推断出的单倍型可以与RNA-seq reads中包含的跨越杂合子SNP的信息相结合,以提高准确性。然而,这对于常见的变异往往是准确的,但对于罕见的变异却不确定。
等位基因特异性reads是许多ASE工具的必要输入。这些reads可以用ASEReadCounter等工具为杂合的SNPs确定。然而,将reads mapping到单倍型而不是单个杂合的SNPs为ASE分析提供更大的力量。使用phASER(RNA-seq的分型和等位基因特异性表达)可以从RNA-seq数据中估计单倍型的表达水平,从RNA-seq reads中获得的单倍型可以使用phASER-pop与群体水平的分型进行整合,将单倍型扩展到未转录区域的假设调控变异(图2c)。一些工具,如IDP-ASE 和BYASE将单倍型作为ASE估计的一部分。对于像EAGLE这样将read counts作为输入的工具,可以提供基因水平的单倍型counts,而不是杂合SNP counts,像phASER每个基因产生一个count。
3.1.4. Considerations for analysis of allele-specific expression in cancer.
在癌症研究中分析等位基因不平衡时,体细胞拷贝数改变(SCNA)可能是一个混杂因素,导致ASE的假阳性。最近的一项泛癌症研究显示,SCNAs占观察到的等位基因不平衡的84.3%。一些研究通过过滤与拷贝数变异重叠的位置来解决这个问题。在评估体细胞突变的等位基因不平衡时,已经开发了考虑拷贝数变异和肿瘤纯度的方法。由于癌症中存在高频率的体细胞突变和拷贝数改变,基因分型通常是基于正常样本的。癌症和正常样本的比较可以揭示在杂合度丢失的情况下保留在癌症中的等位基因,这可以为导致癌症发展的过程提供信息。另外,可以分别估计肿瘤和正常样本的ASE,并比较两组之间显示ASE的SNP的比例。其他研究比较了全外显子组序列和转录组序列中杂合子SNP的变异等位基因频率,或使用基因组DNA中的等位基因比率来校正拷贝数变异的影响。
3.2. Statistical Methods
为分析ASE,人们开发了广泛的统计模型。大体上,它们的特点是,目标是检测单个样本内的等位基因不平衡,或者结合多个样本的数据,以确定ASE的特征,或利用它来帮助估计假定的调控变异的影响(图2)。对于前一个目标,最简单的方法是将mapping到参考(或替代)等位基因的reads作为一个二项式随机变量。一些贝叶斯方法也被提出来分析个体内的ASE。专注于估计ASE的方法可以根据它们是否在单个样本中逐个基因地应用,如二项式检验和一些更专业的方法,或者它们是否试图通过同时考虑多个基因来学习模型参数而加以区分。值得注意的是,当应用于单个样本时,这些方法都不能确认这里定义的ASE,因为通常不可能区分观察到的不平衡是否有遗传来源。已经开发了一些方法,通过将多个个体的表达不平衡与基因型联系起来,推断不平衡的遗传来源。特别值得注意的是开发了一些模型,旨在通过将 ASE 与个体间基因表达水平的变化结合起来,了解调节变异的影响。在此基础上,最近的工作是利用ASE来估计人类基因表达的预期变异,对理解遗传疾病机制有重要意义。虽然本节的重点是分析ASE的方法,但类似的方法也可以应用于其他类型的等位基因变异,如等位基因特异性染色质修饰。
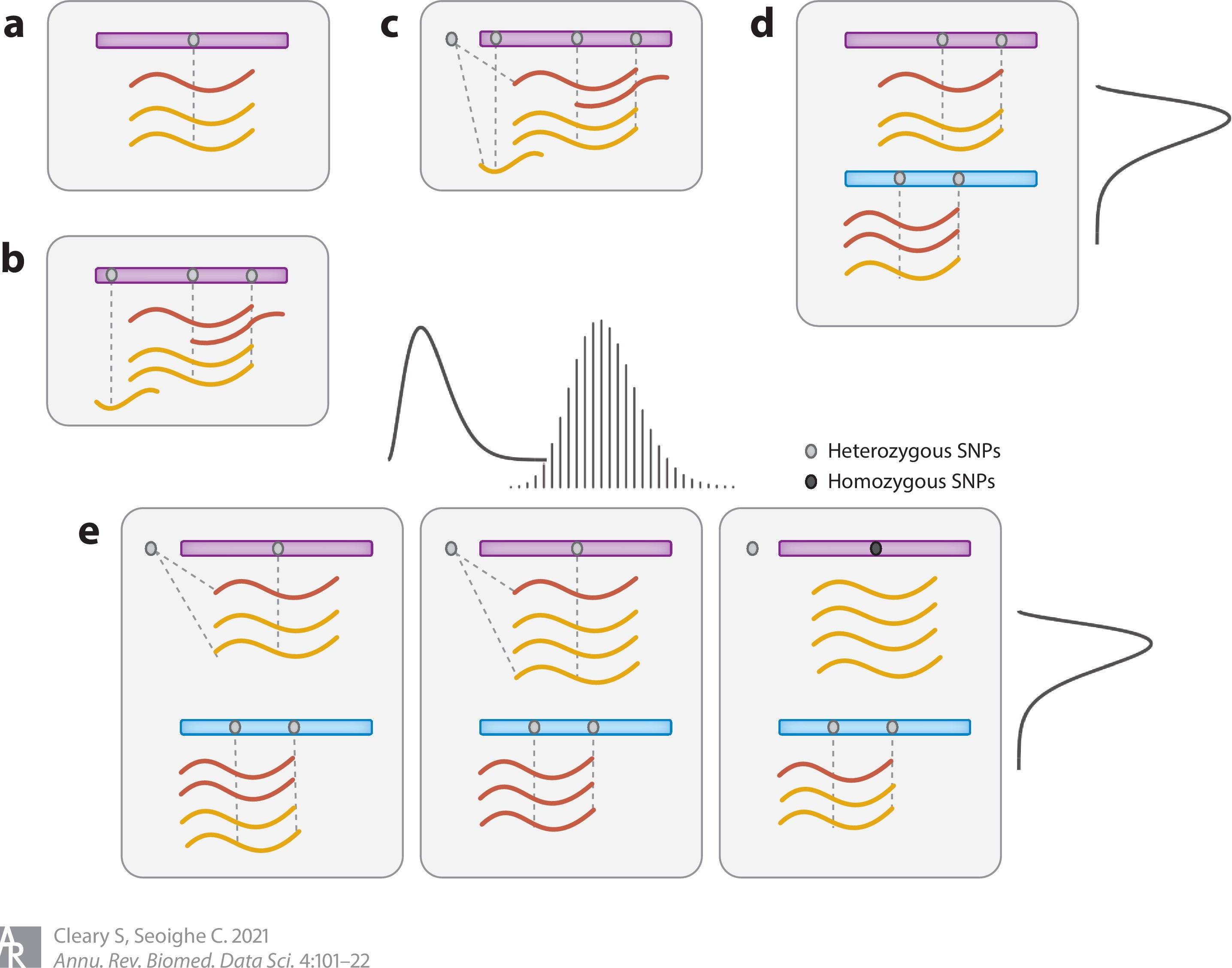
box代表单个等位基因。填充的灰色圆圈代表杂合单核苷酸多态性(SNPs),黑色圆圈代表纯合SNPs。mapping到(a)单一杂合SNP或(b)跨越多个表达杂合SNP的单倍型的等位基因的序列reads可以测试两个等位基因的不平衡的代表性。(c) 当考虑到基于种群的分型时,单倍型可以扩展到假定的调控性SNPs。如果有来自多个个体的数据,这就可以测试表达不平衡的程度和方向与假定的调控性SNP的等位基因的相关性。(d) 统计建模可用于描述单个样本内各基因间等位基因特异性表达(ASE)变化的分布参数。(e) 可以构建模型,将来自杂合子个体的ASE的证据与来自个体间基因表达水平变化的证据结合起来,以检测调控性变异。这些模型包括描述同一基因和不同基因的SNP之间的等位基因表达比率的分布,以及不同个体中的基因总表达水平的分布。
3.2.1. The binomial test and its limitations.
一些早期的等位基因不平衡的研究是基于芯片数据,并采用了基因表达分析和基因分型的方法来比较替代等位基因的表达。当前的研究,使用测序,产生mapping到参考等位基因和替代等位基因的counts(图2a)。这些计数最初是用二项式检验来比较的。应用于单个杂合子SNPs,最直接的方式是提出一个零假设,即随机抽样的序列reads产生于参考或替代等位基因的概率相同。这个零假设可以被修改,以考虑到有利于参考等位基因的mapping偏差,在没有不平衡的零假设下,设定一个略高的read counts产生于参考等位基因的概率。通过将reads mapping到分型的单倍型而不是单个杂合子位点,可以进一步提高功效,RNA-seq reads内的信息,包括等位基因不平衡,可以利用来获得分型的信息,也可用于罕见的变异。还开发了统计模型,用于联合推断杂合子SNPs和检测RNA-seq reads中的ASE。与许多其他推断ASE的方法一样,后一种方法使用似然比检验来评估与替代等位基因的相对应的零假设,同时考虑到推断基因型的不确定性。
使用二项式检验及其变异推断等位基因不平衡,有几个主要的注意事项。相对于二项分布,等位基因的reads数据往往是过度分散的,这意味着mapping到一个等位基因的reads的方差高于二项随机变量的预期。这种过度分散可能既有生物原因,也反映了真实等位基因不平衡的高发率,也有技术原因。可以将来自其中一个等位基因或单倍型的reads作为一个β-binomial[或binomial-logit-normal]而不是二项式随机变量。β-binomial是一个双参数分布,当二项式随机变量的参数本身是一个β分布的随机变量时,就会出现这种分布。它可以用一个平均数和一个过度分散参数进行参数化,后者控制相对于二项式参数的方差增加程度。然而,如果过度分散主要来源于生物学,反映了高频率的等位基因不平衡,那么将从数据中估计的过度分散参数纳入零假设可能会导致检测ASE的能力下降。
过度分散的技术来源之一是重复reads的存在,但这可以通过去除重复的reads来解决,如前所述,或通过使用分子条形码。最近有报道说,技术重复间的等位基因不平衡结果缺乏重现性,并被解释为表明文库制备的其他步骤可能是比PCR扩增更重要的等位基因表达分析的偏差来源。这种缺乏可重复性的情况与早先在Geuvadis研究中从技术重复中得到的结果形成了对比,后者表明在实施质量控制步骤后,各技术重复的方差与二项分布下的预期值相似。等位基因不平衡的假设检验的一个关键缺点是,它把重点放在评估一个零假设上,这可能是不现实的,而且对测序深度敏感,而不是估计等位基因之间的不平衡程度。最后,检测单个个体的等位基因不平衡的方法不容易区分遗传和表观遗传的原因,因此不能用来推断ASE(这里所用的ASE意味着遗传来源)。尽管有上述潜在的局限性,二项式检验仍被用于检测等位基因表达不平衡,也许是由于其易于解释和使用。
3.2.2. Bayesian models for allelic imbalance.
已经开发了几种贝叶斯方法来分析等位基因的不平衡性。考虑到只有一个基因和一个个体的数据,但有多个SNP,IDP-ASE同时从RNA-seq数据中进行单倍型重建和等位基因表达不平衡的推断。采取一个先验,它从重建的单倍型的联合后验概率和随机reads来自个体中的一个或其他单倍型的概率中取样。Skelly等人开发了一个等位基因不平衡的分层贝叶斯模型,同时考虑来自多个基因的数据(图2d)。这首先被用于RNA-seq数据,这些数据来自于酿酒酵母菌菌株的杂交和单一人类细胞系的数据。该研究还包括基因组数据,这使得技术上的误差,如mapping偏差,可以被考虑在内。RNA-seq数据的模型由混合先验组成,其中一个成分对应于等位基因不平衡,另一个成分对应于非等位基因不平衡,其中等位基因的特定reads与基因组数据的分布相同。对于不平衡部分,特定基因中的等位基因特异性reads用β-binomial建模,用期望值和超散性作为参数。在所有基因中,期望值和过度分散本身是β分布的,具有独立的参数,允许基因在杂合子SNP中具有可变或相对稳定的等位基因不平衡。马尔科夫链蒙特卡洛被用来从具有不平衡表达的基因比例、预期值和每个基因的不平衡过度分散的联合后验分布中获得样本,以及描述这些在具有等位基因不平衡的基因中如何变化的参数。这种贝叶斯方法的优点是能够对受等位基因不平衡影响的基因的总体比例和这些基因的效应大小分布作出推断。同一小组使用了混合效应二项式回归模型的贝叶斯方法,结合不同个体和不同组织的信息来估计与尼安德特人引入相关的ASE。描述非参考reads数量的二项分布参数被建模为固定截距项(对应于ASE效应)与组织和个体的随机效应之和。最近,Dong等人开发了一个贝叶斯模型和一个Python库,以估计任何倍性>1的基因和同源物水平的表达不平衡。然而,据我们所知,还没有进行独立的基准测试来评估这些方法的性能。
3.2.3. Combining allele-specific expression and expression level to estimate regulatory effects of genetic variants.
当有来自多个个体的数据时,就有可能通过确定不平衡和附近的推定调控变体之间的关联,来推断观察到的等位基因不平衡(即本综述中定义的ASE)的遗传原因。当单倍型可以扩展到转录区以外,包括调控区的推定致病变体时,这一目标就会得到加强。除了检测低频率的调控变异的高能力外,将等位基因不平衡与已知的顺式eQTL联系起来,可以确认与受影响基因共同定位的eQTL是顺式作用的。一个顺式作用的调控变异有能力改变整个样本的基因表达水平,并改变变体为杂合子的单个样本中等位基因的相对表达水平。在没有负反馈回路的情况下,在某些情况下可能会缓冲调节性变异的影响,调节性变异对不同样本的基因表达水平的影响和相关等位基因不平衡的程度之间存在着直接的关系。Mohammadi等人将等位基因倍数变化(aFC)定义为杂合子个体中相对于参考等位基因的替代表达。在一个与基因型有关的跨个体的基因表达的线性模型中(对参考同源者、杂合子和替代等位基因同源者分别编码为0、1和2),aFC对应于β 1 / β 0 + 1,其中β 0是截距,β 1是对替代等位基因每拷贝的基因表达的影响。
如果目标是利用ASE来了解调控变异对表达的影响,最好是对基因表达水平和调控变异的等位基因不平衡效应进行联合建模,而不是分别估计ASE和eQTL效应(图2e)。这样,ASE补充并大大增强了识别顺式作用的eQTLs的能力。一些类似的统计模型已被应用于此目的。WASP中的组合单倍型测试(CHT)以及RASQUAL(稳健的等位基因特异性定量和质量控制)和TReCASE(总读数ASE)的模型涉及一个基于个体间映射到感兴趣基因的读数数量和个体内映射到特定单倍型的读数联合概率的似然函数。这三种方法都使用似然比测试来检验基因对表达的影响。跨个体的读数被建模为负二项式(或在CHT的情况下为负β二项式),等位基因特定的读数则使用β二项式建模。负二项在RNA-seq分析的早期方法中被用作Poisson分布的替代品,以允许在样本间观察到读数的超额方差,类似于上文讨论的贝塔二项被用作二项分布的过度分散替代品的方式。这些方法的细节有一些不同,特别是过度分散的建模方式。例如,CHT对读深度使用基因和样本级的综合过度分散参数,而RASQUAL使用单一的基因特定参数来模拟总读数和等位基因特定计数的过度分散。Kumasaka等人的补充表2总结了建模选择的其他差异,其中包括对基因分型和相位的不确定性的处理。这些作者报告说,与CHT和TReCASE相比,他们的RASQUAL方法具有更高的功率;但是,据我们所知,对这些和相关任务的模型和工具的性能也没有进行全面和公正的评估。
3.2.4. The expected variance in gene expression.
上面讨论的大多数方法的目的是估计不同基因等位基因的相对表达。Mohammadi等人最近开发了一种方法,其目的不是利用ASE,而是为了估计基因表达的预期变异,VG与作用于顺式的所有调控变体集合有关。该模型假定杂合转录的SNP与未观察到的双列调控变体存在不完全的联系。它还允许存在大量其他影响较小的顺式调控变体,并援引中心极限定理得出转录变体的参考等位基因所产生的读数的二元正态分布。这个分布类似于上文讨论的经常被用来模拟ASE的β-二项式随机变量。以这种方式估计的基因表达的遗传变异具有基本的科学意义,它提供了关于作用于基因表达的选择约束的信息,并与理解基因表达如何随时间演变有关,但它也提供了一种对候选疾病相关基因和突变进行优先排序的手段。该方法被称为表达变异分析(ANEVA),可用于加强对表达异常值的检测,通过突出具有正常约束基因表达的基因的表达异常值。ANEVA与剂量离群值测试的结合(称为ANEVA-DOT)被应用于孟德尔肌肉萎缩症和肌病患者,并被证明有很高的能力来恢复已知的因果变异,以及提出新的潜在因果变异,其中一个变异在研究中被证实。总的来说,这个ASE的统计模型和受其启发的其他模型可能成为诊断遗传病因果变异这一关键问题的分析工具库的重要组成部分。
3.2.5. Single-cell data and cell type–specific imbalance.
Bulk RNA-seq数据来自于通常由多种细胞类型组成的样品,以不同的比例混合。基因在组成细胞类型中的调节方式不同,导致不平衡程度的变化取决于细胞类型的组成。这可以通过首先按细胞类型分类后对单细胞RNA-seq数据进行等位基因不平衡分析来解决。然而,产生适当的单细胞数据仍然是昂贵和具有挑战性的,迄今为止,对多个个体组织的基因表达的大规模调查只涉及大量的RNA测序。基因表达去卷积方法可用于估计Bulk RNA-seq数据中的细胞类型比例,特别是当组成细胞类型的基因表达可从相关的单细胞实验中获得时。然后可以与不平衡分析联系起来,推断受等位基因不平衡影响的细胞类型。BSCET(bulk and single-cell RNAseq data to detect cell-type-specific allelic expression imbalance)方法利用等位基因不平衡的程度和整个样本的细胞类型比例之间的统计学互动来推断受bulk RNA-seq数据中等位基因不平衡影响的细胞类型。
4. CONCLUSIONS
等位基因不平衡在人类基因的各个层面都很常见,从染色质状态到mRNA表达水平和剪接以及蛋白质翻译的速度。大规模研究的证据表明,大多数基因表达的等位基因不平衡是由遗传引起的。基因表达的遗传不平衡,在此称为ASE,对疾病风险和致病编码序列变异的表型影响的严重程度有影响。二倍体个体的细胞提供了关于调控变异影响的读取,使人们能够探索假定的调控变异的功能后果,甚至在罕见变异的情况下。大量的计算方法和统计模型已经被开发出来,以评估个体内和个体间的等位基因不平衡所提供的信息,后一种方法通常能够将ASE与表达水平的个体间差异相结合,以评估调控变异的影响。对这些方法进行进一步的独立基准测试将有助于指导最佳分析。随着检测人类群体中对人类表型变异有细微影响的遗传位点的能力越来越强,对能在分子水平上准确推断基因组变异影响的方法的需求也越来越大。此外,还需要改进方法来评估疑似参与疾病的新发变体的可能后果。利用ASE来帮助评估正常人群中基因表达的变异,以优先考虑对基因表达有较大影响的变异,相对于人群中的变异,这是一个特别有希望的方向,它强调了ASE对疾病相关研究的作用。
5. Reference
https://doi.org/10.1146/annurev-biodatasci-021621-122219